ExaBiome Brings Metagenomics into the Exascale Era
By Linda Vu, Lawrence Berkeley National Laboratory

The Exabiome project’s Principal Investigator Kathy Yelick and Executive Director Leonid Oliker. Photo credit: Margie Wylie, Berkeley Lab
Microorganisms are essential for life: they play a vital role in modulating and maintaining our atmosphere, supporting plant and animal growth, keeping humans healthy, and much more. By understanding how they work, researchers could discover solutions to challenging problems in biomedicine, agriculture, and environmental stewardship.
Until recently, a significant hurdle to understanding how microbes sustain life is that they don’t work alone. Instead, they function in nature as dense, complex communities that are often physically or metabolically interconnected. In the last 20 years, the field of metagenomics has allowed scientists to collectively sequence the genomics of these microbial communities on their own “turf” to unlock the mystery of how they work.
But these metagenomic datasets are massive, and before now, researchers lacked the computational power and software tools to analyze them effectively. The Exabiome project, led by Lawrence Berkeley National Laboratory’s (Berkeley Lab’s) Katherine Yelick and Leonid Oliker, helped to bridge this gap by developing novel software tools that allow researchers to harness the power of cutting-edge high performance computers (and now exascale supercomputers) to solve previously infeasible problems in metagenomics. And partnerships with the Department of Energy (DOE) Joint Genome Institute (JGI), Los Alamos National Laboratory, and Indiana University enabled the team to achieve transformative work.
“Overall, we’ve been able to analyze datasets that would have been impossible before we built these tools. Exabiome’s work allows computational biologists to take advantage of high performance computing (HPC) systems, for which these tools didn’t previously exist,” said Yelick, Exabiome’s principal investigator. “This effort, combined with advances in exascale computing, will allow these researchers to do very large-scale analyses at a much more regular pace.”
“As these tools become available, researchers are bringing forth bigger and bigger datasets. Many of them didn’t realize that the technology existed to analyze this volume of data,” said Oliker, executive director of the Exabiome Project. “So now we’re in a chicken and egg situation: As people realize that we can do large metagenomic assemblies, they’re bringing us even bigger samples for analysis. And as these enormous samples become available, we can push the state-of-the-art in computational biology even further.”
Transforming Metagenomic Data Analysis
Initially, the Exabiome project set out to develop scalable software tools for three core computational problems in metagenomic data analysis: metagenome assembly, protein clustering, and comparative metagenome analysis.
According to Yelick, the team achieved all these goals over the past five years and even tackled a fourth challenge: using machine learning, particularly graph neural networks, to understand the functional characteristics of proteins in metagenomic samples. Here are some ways that Exabiome’s tools have transformed metagenomic data analysis.
MetaHipMer: Metagenome Coassembly
After collecting an environmental sample, scientists will typically run it through a gene sequencer to gain insights into the makeup of the microbial community. The machine will produce fragments of DNA sequences, which include the DNA of every microbe in the sample all jumbled together. Researchers must reassemble the genome to identify individual microbes in the community and determine their functions.
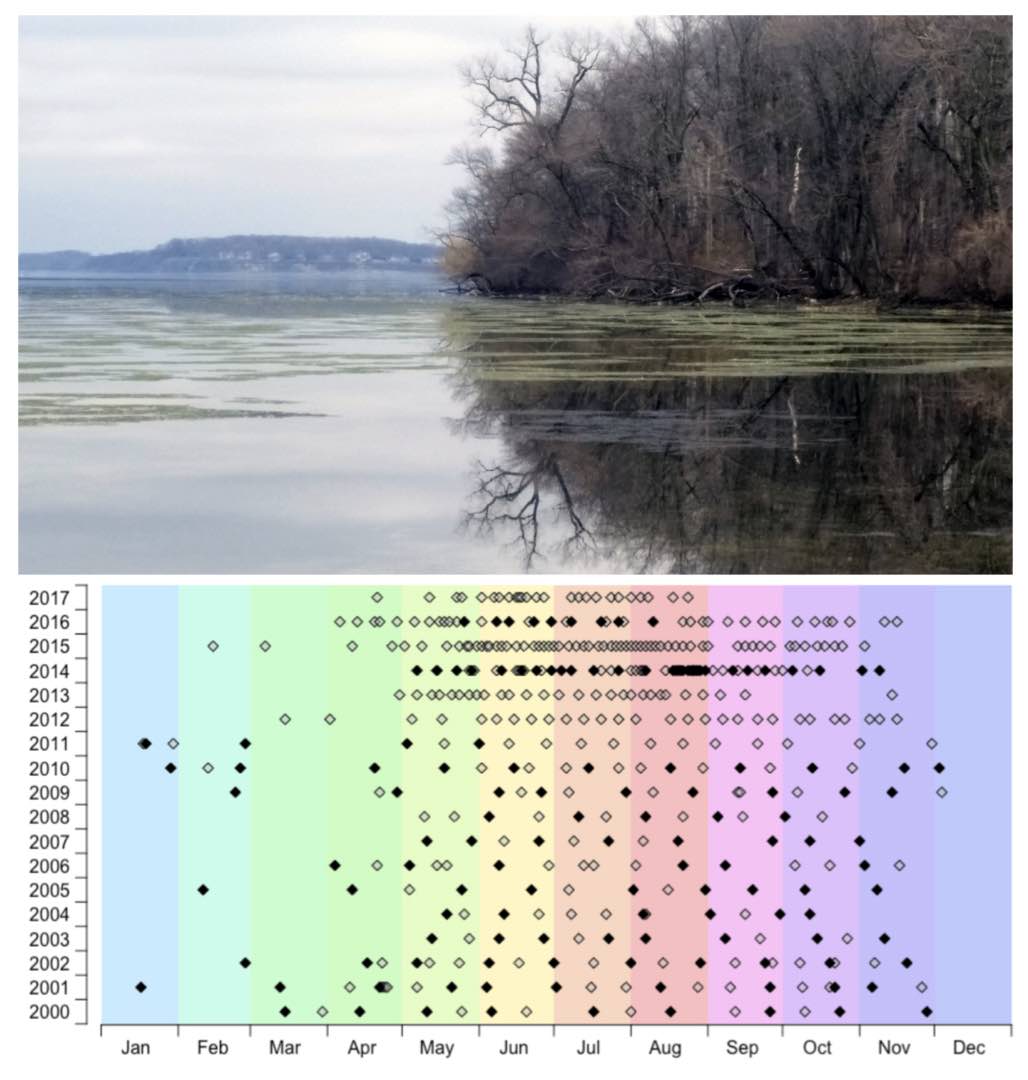
Top: MetaHipMer2 is being used to support a broad array of metagenomics-based user science at the JGI. One of the datasets involves samples from Lake Mendota, a large freshwater lake in Wisconsin. Photo credit: Trina McMahon Lab. Bottom: Time series graph with sample dates in the Lake Mendota archive. Filled symbols correspond to dates for which 16S rRNA gene tag data are already available. Image credit: Trina McMahon Lab, JGI 2019 Community Science Program proposal
Yelick and Oliker compare this process to putting a book through a paper shredder and then rebuilding the sentences, paragraphs, and chapters from text snippets. And just as strips of paper may get mangled in the “shredding” process, the sequencing process can introduce similar errors.
“If I’m only shredding one book, it’s going to be hard to put the book back together because I don’t know where the errors are. But if I shred 50 copies of the same book, it’ll be easier to piece the book back together because I’ll have 50 copies to compare it to, and it’s unlikely that errors will occur in the same place,” said Yelick.
It’s the same with metagenomes, she added, which is why analyzing these datasets requires so much computer memory. Depending on the complexity of the data, Yelick says the high watermark for computer memory needed to do the analysis could be five or ten times the size of the raw data input. So if a dataset like Tara Oceans, the biggest genetic sequencing task ever undertaken on marine organisms, is about 100 terabytes of raw input data, it could require up to a petabyte of data to complete the analysis.
Before Exabiome’s contributions, metagenomic assembly tools primarily relied on shared memory, which is one block of random access memory used by several different processors. This limitation restricted the size of the metagenomes that researchers could assemble. As a result, they resorted to multiassembly—assembling smaller samples on shared memory and then combining the results into a big picture.
Because the Exabiome team designed its MetaHipMer assembly tool to rely on distributed memory—where each processor has its private memory and data is passed amongst processors—researchers are no longer constrained by limited memory. They can scale up to thousands of nodes on the Summit supercomputer and coassemble terabyte-size metagenomic datasets all at once instead of combining the results from multiple partial assemblies. Oliker notes that MetaHipMer can also complete these assemblies fairly rapidly, on the order of minutes or hours for multiple terabytes of data.
“It’s not just about handling bigger datasets; we’ve shown that the bigger datasets give you better science results,” said Yelick. “When you analyze large metagenomic datasets as a whole instead of in smaller chunks, you get more microbial species, especially rare species, and longer contiguous information about the metagenome’s (or microbial community’s) makeup.”
HipMCL and PASTIS: Clustering at Scale
After the assembly process, researchers want to identify patterns and relationships between microorganisms that could point to structural and functional similarities. To do this, they rely on clustering algorithms. This process essentially groups objects so that items in the same group (cluster) are more similar than those in other clusters.
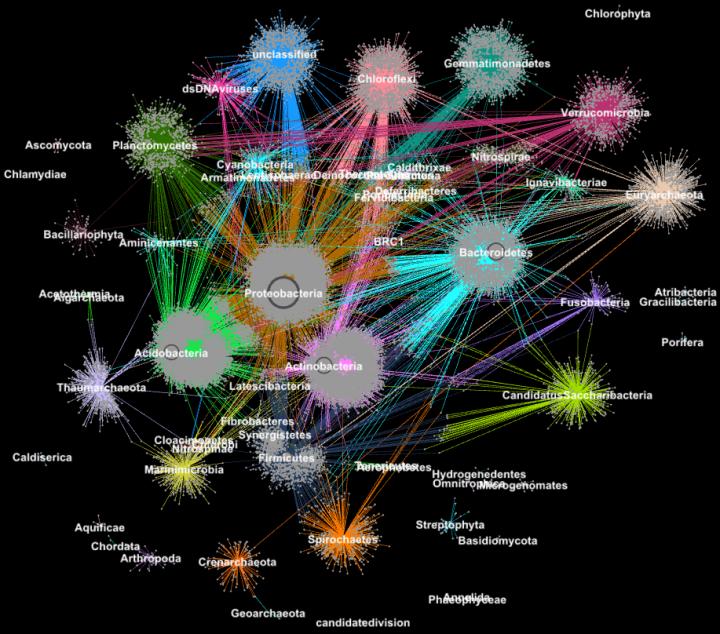
Proteins from metagenomes clustered into families according to their taxonomic classification. Image credit: Georgios Pavlopoulos and Nikos Kyrpides, JGI/Berkeley Lab
Before Exabiome, computational biologists had several go-to techniques for clustering—the most popular was the Markov Clustering (MCL). But as advancements in high-throughput sequencing technologies led to explosive growth in data, many of these techniques encountered a computing bottleneck. In fact, very few of these algorithms could cluster a biological network containing millions of proteins (nodes) and connections (edges).
For instance, MCL mostly ran on a single computer node, was computationally expensive to execute, and had a big memory footprint—all of which limited the amount of data this algorithm could cluster. So the Exabiome team modified MCL to run quickly, efficiently, and at scale on distributed-memory supercomputers. The team even developed state-of-the-art algorithms for sparse matrix manipulation, which enabled the tool’s scalability. They called this tool HipMCL.
In an early test run, HipMCL achieved a previously impossible feat: clustering an extensive biological network containing about 70 million nodes (proteins) and 68 billion edges (connections) in about 2.5 hours using approximately 140,000 processor cores on the National Energy Research Scientific Computing Center’s (NERSC) Cori supercomputer.
Working closely with the ExaGraph Co-Design Center, the Exabiome team also developed Protein Alignment via Sparse Matrices (PASTIS), a distributed memory software for large-scale protein similarity searches. PASTIS constructs similarity graphs from extensive collections of protein sequences, which a graph clustering algorithm can use to discover protein families. PASTIS enhances its scalability by using distributed sparse matrices as part of its underlying data structure.
In a recent run, the team used PASTIS to perform an all-vs-all protein similarity search on one of the largest publicly available datasets with 405 million proteins in less than 3.5 hours using 20,000 GPUs on the Summit supercomputer. Their paper describing this work, “Extreme-scale many-against-many protein similarity search,” is a finalist for the prestigious Gordon Bell Prize. The winner will be announced at SC22 in Dallas this November.
“PASTIS did this unprecedented protein similarity search two orders of magnitude faster than the current state-of-the-art; we’re talking about reducing the analysis time from months to hours,” said Oliker. “Our work is opening the door for these types of massive protein analyses. We have several papers in review at prominent scientific journals citing this tool. Our goal now is to do a protein similarity search on 500 million proteins and more on an exascale system. “
Kmerprof: Comparing Metagenomes
Leveraging some of the software they built for assembly, Yelick notes that the Exabiome team also developed a high performance scalable tool called kmerprof for comparing one metagenome to another.
“This tool allows you to look at the collected data and say what’s different and what’s the same,” said Yelick. “One of the researchers we’re collaborating with at JGI is looking at how the community of bacteria, fungus, algae, etc. has changed in Wisconsin’s Lake Mendota over 50 years. Our tool allows them to compare samples collected over seasons and years to see what’s changed.”
Researchers could also use this tool to compare metagenome samples from human guts worldwide to better understand how diets and environments impact our microbiota and health, she noted. The possibilities are endless.
Exascale, Machine Learning, and Beyond
Although machine learning wasn’t something the Exabiome team planned to work on when the project began, the team is exploring these tools as part of other smaller collaborations. They are particularly interested in using graph neural networks for taxonomic classification and factorization-based community structure detection, among other things.
“Machine learning will grow to be more important as we try to understand the functional characteristics of these proteins,” said Yelick. “Like in many areas of scientific computing, there are tremendous opportunities for machine learning in computational biology. We’ve only scratched the surface in exploring some of these avenues.”
Looking back, Oliker considers the Exabiome team’s ability to leverage graphic processing units (GPUs) to perform large-scale metagenomic assemblies one of the team’s most significant challenges and accomplishments.
“I don’t think people realize that performing large-scale assemblies is a significantly different HPC problem than almost any other application area within the Exascale Computing Project. There are a lot of string manipulations, distributed hash tables, and no floating point operations, which creates unique challenges when we had to think about porting our software onto GPUs,” said Oliker.
He adds, “at first, we were concerned that with the unique algorithmic structure of our codes, the load balancing issues, and the string manipulations, we wouldn’t be able to leverage GPUs effectively. But our team has been surprisingly and impressively successful at finding novel ways of accelerating these things on GPU architectures. Seeing their resourcefulness has been a really exciting part of this project.”
In fact, the Supercomputing (SC) Conference selected one of the team’s solutions, “Accelerating Large Scale de Novo Metagenome Assembly Using GPUs,” as the best paper finalist at SC21. Another Exabiome achievement, “Extreme scale de novo metagenome assembly,” was also selected as the best paper finalist at SC18. And HPCwire recognized the team’s resourcefulness with a Reader’s Choice Award in 2015 and an Editor’s Choice Award in 2021 for the best use of HPC in life sciences.
The Exabiome team is now seeking collaborations to analyze some of the world’s largest metagenomic datasets, such as Tara Oceans, to showcase their tools’ capabilities. Marine biologists collected the Tara Oceans data from nearly every sea on the planet over four years. The Exabiome team also hopes to analyze the entire human microbiome dataset collected from the globe.
“These are exciting collaborations that will enable new scientific insights and hopefully inspire people to bring even more new data for us to analyze, to help uncover the unknown,” said Oliker.