By Rob Farber, contributing writer
The ExaBiome project has been selected as a 2022 Gordon Bell Finalist. The prestigious ACM Gordon Bell Prize is awarded each year at the International Conference for High Performance Computing, Networking, Storage, and Analysis (SC) to recognize outstanding achievement in high-performance computing (HPC) and reward innovation in applying HPC to applications in science, engineering, and large-scale data analytics.
Kathy Yelick, PI of the Exascale Computing Project’s (ECP’s) ExaBiome effort and Vice Chancellor for Research at UC Berkeley, highlights her team’s vision about this project and its future, “The ExaBiome team is building a toolset to work with terabytes of data (containing trillions of base pairs) where we don’t understand all the proteins coded for by these base pairs at this time. Understanding the similarities between proteins and their interrelationships is important to understanding how microbiomes work. To do so requires very powerful computational tools that can infer the functions of related proteins in these massive datasets.” Microbiomes are cooperative communities of microbes. These organisms and the organisms in associated microbiomes are central players in climate change, environmental remediation, food production, and human health (Figure 1).[1]

Figure 1. Microbiomes are critical to energy production and the environment.
Focusing on the importance of the ExaBiome research performed for the Gordon Bell competition, Ananth Kalyanaraman (professor, Boeing Centennial Chair at the School of EECS in Washington State University, and Director of the AgAID Institute) explains the importance of understanding protein similarity, “Protein sequence similarity search (or homology search) is a problem of foundational importance to molecular biology. The problem is one of identifying similarities between protein sequences. Similarities at the sequence level often point to structural and hence functional similarities. Consequently, homology searches are often one of the very first steps in bioinformatics pipelines.”
Srinivas Aluru (professor in the School of Computational Science and Engineering and executive director of the Institute for Data Engineering at Science Georgia Institute of Technology) recognized the challenge in achieving Gordon Bell levels of performance on protein homology searches in his review of the research, “Identifying homology from protein sequence is a highly computationally demanding task. Conceptually, one has to compare all protein pairs to each other to discover their average nucleotide identity (ANI). Unfortunately, this is not feasible with 100s of millions of proteins on novel environmental samples, as that would translate into O(1016 to 10 18) pairwise protein alignments, each of which take time proportional to the product of the two sequences aligned.”
To demonstrate the success of their approach, the ExaBiome team utilized over 20,000 GPUs on Oak Ridge National Laboratory’s (ORNL’s) Summit supercomputer to perform a many-against-many protein similarity search on the largest available protein dataset. The performance observed was transformative because the search completed in hours rather than in weeks. More specifically, the paper, “Extreme-Scale Many-Against-Many Protein Similarity Search,” reports a new high water mark for performance by achieving a search in 3.4 hours on a large, publicly available dataset with 405 million proteins.[2], [3]
Aydın Buluç (senior scientist at Berkeley Lab and adjunct assistant professor of EECS at UC Berkeley) notes that the many-against-many search performance tells only part of the story because the speed of data generation and memory consumption also presented computational challenges.
- Going from raw sequencing data to protein similarity networks: According to the Gordon Bell submission paper, “Clustering proteins is important to understanding which proteins are evolutionarily and functionally related. The challenge is that the HipMCL network clustering algorithm was so fast that the generation of the input network became a bottleneck. This is why we developed the Protein Alignment via Sparse Matrices (PASTIS) tool to generate protein similarity matrices equally fast.”[4]
- Reducing memory consumption: An innovative matrix-based blocking technique was also developed to address “a huge memory footprint that makes it hard to scale the problem sizes.”[5]
The protein family identification pipeline (Figure 2) addresses these computational challenges and, along with the use of GPU acceleration and MPI distributed computing, has given computational biologists a transformative advance in capability. The authors note that, “overall, we increase the scale of the solved problem by an order of magnitude (15.0×) and improve the performed alignments per second by more than two orders of magnitude (575.5× ).”[6] The pipeline scales well, which means even more alignments per second can be achieved on larger computers—thus greatly expanding the opportunities in metagenomic research and comparative biology in general.
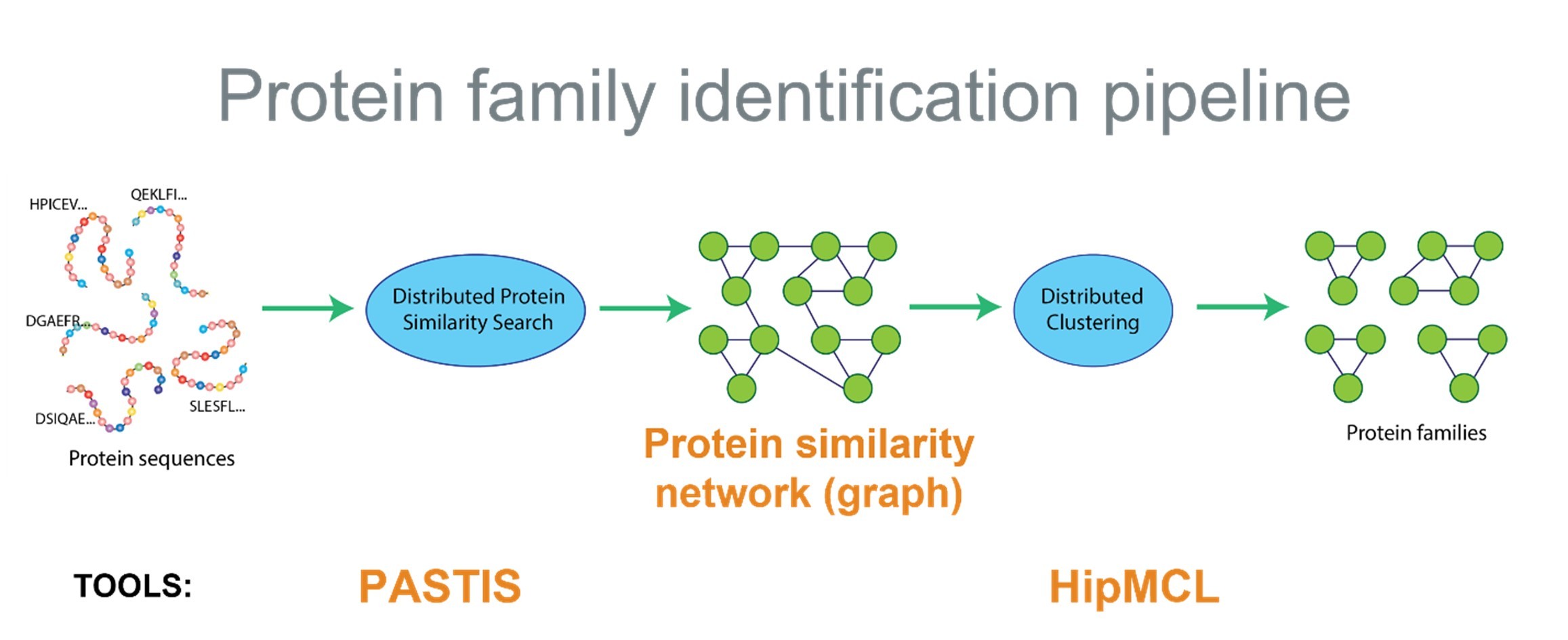
Figure 2. The protein family identification pipeline (Source: ExaGraph team).
A Multiyear, Multi-Organizational Effort
The Gordon Bell submission is the fruit of a multiyear, multi-organizational effort spearheaded by Kathy Yelik and the Lawrence Berkeley National Laboratory (LBNL) team (Figure 3). The project benefits from the contributions of many individuals and organizations (see the ExaBiome site).

Figure 3. Members of the LBNL ExaBiome Gordon Bell team: Kathy Yelik, PI (top left); Lenny Oliker, executive director (top right); Aydin Buluç, senior scientist at Berkeley Lab (bottom left); and Oğuz Selvitopi, research scientist at Berkeley Lab (bottom right).
Several organizations participated in this Gordon Bell submission:
- Applied Mathematics and Computational Research Division, LBNL, USA
- Microsoft Corporation
- University of California, Berkeley, USA
- National Energy Research Scientific Computing Center, LBNL, USA
- Institute for Fundamental Biomedical Research, BSRC, Vari, Greece
- Indiana University, USA
- Joint Genome Institute, LBNL, USA
The team is grateful for contributions from these and other partners including LBNL, the Oak Ridge Leadership Computing Facility, and DOE’s Joint Genome Institute. The ExaBiome team has also benefitted from the students who worked on the Gordon Bell submission, and happily the benefit is mutual. Oğuz Selvitopi (research scientist in the Performance and Algorithms group of Computer Science Department at LBNL) notes that one postdoc who worked on this code received an appointment in computational research. For those students interested in working on the ExaBiome project, please contact the ExaBiome team.
An Exascale Foundation for a Fundamentally Important Molecular Biology Tool
As a part of the ECP, the ExaBiome project benefitted from funding and interactions concerning scalable graph and sparse matrix algorithms with the ExaGraph co-design center led by Pacific Northwest National Laboratory.
Lenny Oliker (executive director of the ExaBiome project, senior scientist, and group lead of the LBNL performance and algorithms group) points out that, “PASTIS is the main code for the ExaBiome Gordon Bell submission developed jointly by ExaBiome and ExaGraph. We can compare ½ billion to ½ billion proteins because the algorithm intelligently uses sparsity.”
Aydin Buluç described the collaborative nature of the work performed for the Gordon Bell competition, “We collaborated with the ExaGraph co-design center a few years ago. The discussions of scalable algorithms helped us create our unprecedented scalable implementation for the Gordon Bell submission.”
Achieving the current levels of performance proved to be a lengthy effort, and the ExaBiome team has been developing the parallel many-against-many codebase for comparative biological research since 2018.
The accuracy of the method used in the SC22 submission was initially established in a 2002 study.[7] Parallelization efforts in 2020 increased performance[8] with the latest incarnation achieving Gordon Bell Award levels of performance. As Srinivas Aluru wrote, “This Gordon Bell nomination increases the sizes of problems that could be solved by the PASTIS implementation by almost 2 orders of magnitude over the problems in the SC20 paper thanks to memory-consumption optimizations, new parallel algorithms that leverage the symmetry in the sequence similarity matrix, GPU acceleration, and the ability to address load imbalance issues.”
The team is looking forward to running on production exascale hardware. Buluç observes, “The ECP has been very helpful in terms of training, including giving us access to the ORNL Crusher supercomputer, which contains identical hardware and similar software as the Frontier exascale system.” Crusher is used as an early access test bed for the Center for Accelerated Application Readiness, ECP teams, National Center for Computational Sciences staff, and vendor partners. The system has two cabinets—one with 128 compute nodes and the second with 64 compute nodes, for a total of 192 compute nodes.[9] Buluç continues, “We believe that we should run efficiently on the forthcoming exascale systems.”
The ECP has been very helpful in terms of training, including giving us access to Crusher, which contains identical hardware and similar software as the Frontier exascale system. We believe that we should run efficiently on the forthcoming exascale systems. – Aydin Buluç
Selvitopi expands on this thought, “We could already perform a distributed search on CPU-based HPC systems. To increase the scale of the problems we can solve and to improve search speed, we focused on leveraging the accelerators. This helped us attain a scale worthy of a Gordon Bell submission. Currently, our tool can run on both AMD and NVIDIA GPUs, and we are looking forward to testing on the Intel GPUs.”
We could already perform a distributed search on CPU-based HPC systems. To increase the scale of the problems we can solve and to improve search speed, we focused on leveraging the accelerators. This helped us attain a scale worthy of a Gordon Bell submission. Currently, our tool can run on both AMD and NVIDIA GPUs, and we are looking forward to testing on the Intel GPUs. – Oğuz Selvitopi
Technology Introduction
Buluç explains the design goal for the Gordon Bell submission as follows: “Scientifically, we want to focus on the relationship between proteins (and the genes that code for those proteins) because this is important to understanding what they do. Clustering gives us a way to see these relationships. The challenge is that the clustering algorithm is so fast that the generation of the input for the clustering algorithm became a bottleneck. To address both the memory and run-time challenges, we created PASTIS.”
Memory utilization per node is a critical challenge, and the paper notes that the number of candidate pairs that must be stored and aligned grows quadratically with the number of sequences in the search, which makes the similarity search over huge datasets even more challenging in terms of memory requirements.[10] Computer scientists express this type of growth in terms of Big-O notation. Thus, quadratic growth is proportional to O(N2), where N is the number of sequences in the search.
Special sparse matrix operators were devised to perform the necessary computations in PASTIS. These sparse matrix data structures, along with innovative algorithm design, reduced the per-node memory usage. According to the paper, the basic information storage and manipulation medium used by PASTIS is sparse matrices. They are used to represent different types of information required throughout the search. Sparse matrices are very common and widely used in linear algebra. Similarly, they can be used to represent graphs, albeit with different sparse matrix operators.[11] The paper notes, “For example, in PASTIS, the discovery of candidate pairwise sequences is expressed through an overloaded sparse matrix by sparse matrix multiplication, in which the elements involved are custom data types, and the conventional multiply-add operation is overloaded with custom operators known as semirings.” Semiring algebra allows PASTIS to express computations in a similarity search through sparse operations (Figure 4).
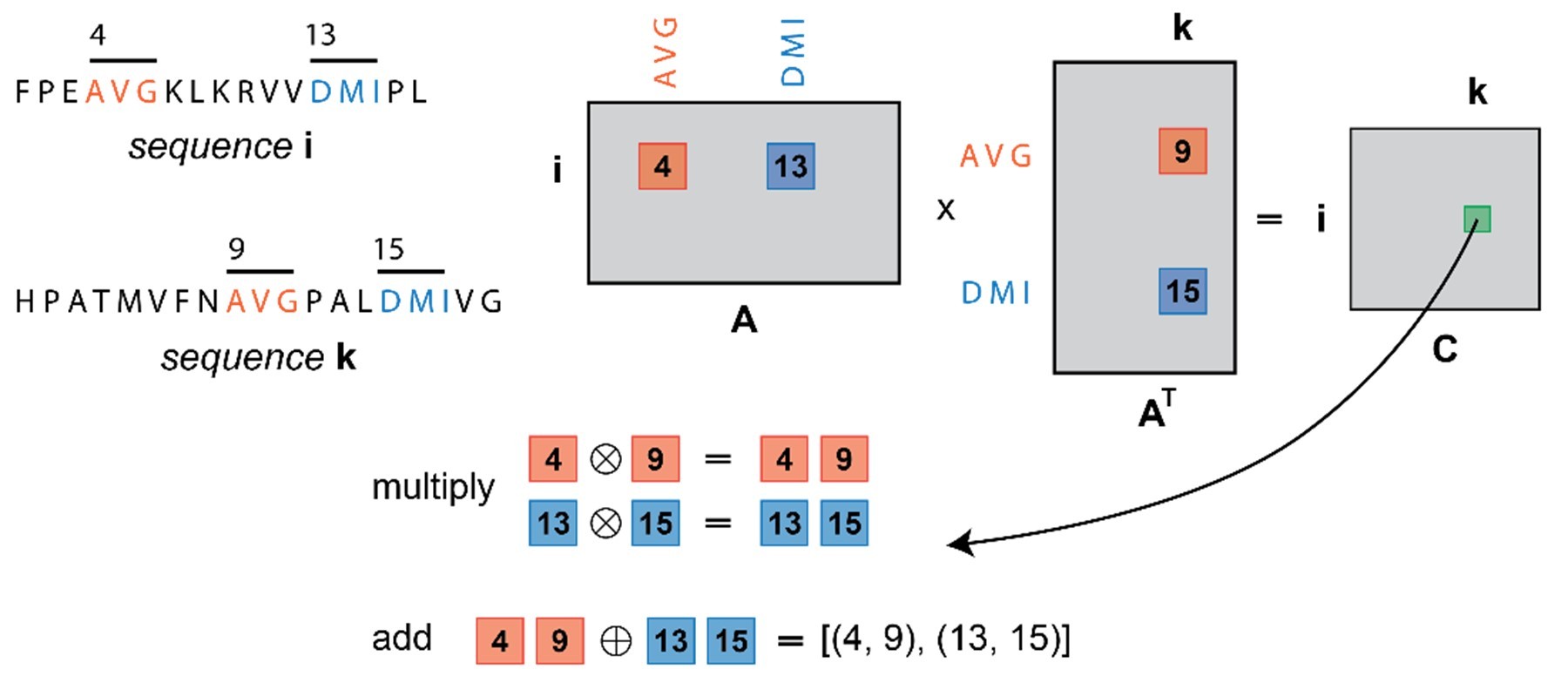
Figure 4. A simple example to illustrate the discovery of a candidate pair for alignment.
The protein similarity search pipeline (Figure 2) utilizes several libraries and orchestrates them in a distributed setting. These libraries are included in the PASTIS GitHub repository.
- CombBLAS is a distributed-memory parallel graph library based on arbitrary user-defined semirings on sparse matrices and vectors.
- SeqAn is an open-source C++ library of efficient algorithms and data structures for analyzing sequences with the focus on biological data. SeqAn supports node-level shared-memory parallelism with vectorization.
- ADEPT is a “novel domain-independent parallelization strategy that optimizes the Smith-Waterman algorithm for DNA and protein sequencing on the heterogeneous architectures and GPUs of petascale supercomputers.”[12] Oliker notes, “The ADEPT library for pair-wise alignment is used in multiple genomics projects, including the code that is the subject of the Gordon Bell submission.”
Apart from these libraries, PASTIS itself uses MPI/OpenMP hybrid parallelism and MPI I/O. Both CPU and GPU resources are concurrently utilized to maximize performance. The PASTIS software stack is shown in Figure 5.
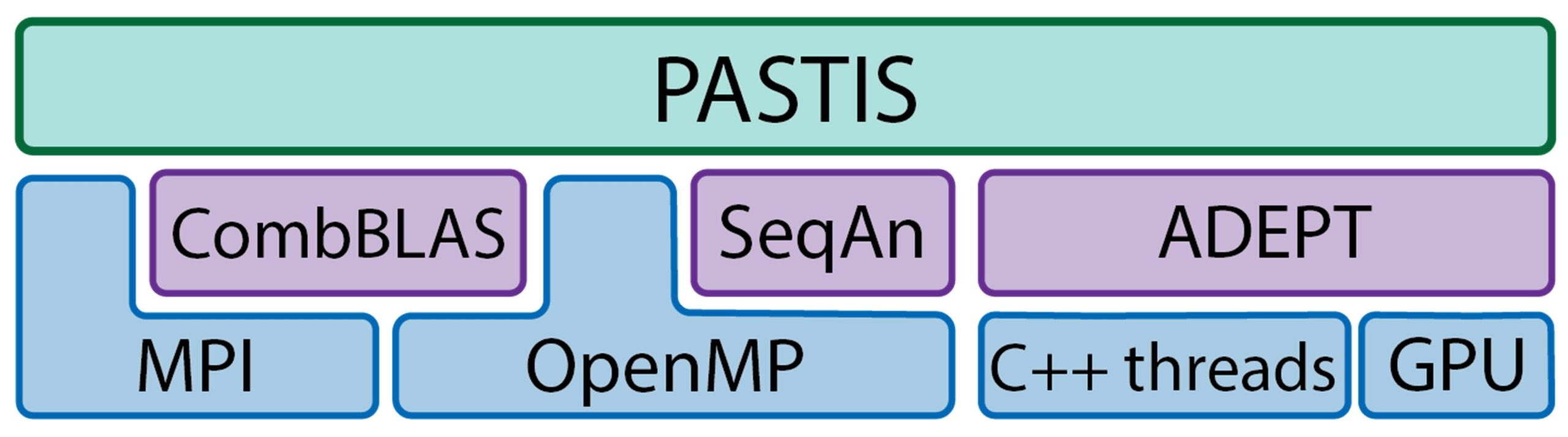
Figure 5. PASTIS utilizes different libraries and can efficiently use both CPU and GPU resources on a node.
Extreme-Scale Protein–Sequence Similarity Search is Now Possible
PASTIS makes extreme-scale protein–sequence similarity search possible by leveraging state-of-the-art HPC hardware. Similarity search is the backbone of bioinformatics, and many important biological tasks depend on the analysis of similar proteins. The larger the dataset being examined, the more thorough the analysis performed by the scientist. Obtaining results in hours rather than weeks when using these large datasets means that scientists can perform previously difficult or intractable analyses on extreme-scale sequence data.
The size and performance numbers tell the story, but ultimately it is the sensitivity and time-to-solution that matters. In this context, sensitivity equates to reporting all or as many of the true positives as possible.[13]
The team notes there are many protein similarity search tools in the literature, and each utilizes different search techniques that have been refined over the years. Some of the more popular tools include BLASTP, MMSeqs2, LAST, DIAMOND , and USearch. In terms of the current state-of-the-art, the authors examined MMSeqs2 and DIAMOND in depth because of the tools’ distributed memory support.
By pressing the limits of software technology, unexpected challenges can occur when attempting to perform exact comparisons using the same datasets. Owing to errors and slower than expected performance (as detailed in the paper), the team ultimately used the published performance results reported by the DIAMOND team, which used the Cobra system at the Max Planck Society[14] for their comparative analysis.
Search Space and Sequence Queried Per Second
The DIAMOND software has user-selectable sensitivity settings designed to balance run time against sensitivity. The DIAMOND team reported that they could perform a search of the National Center for Biotechnology information non-redundant database, which contains approximately 280 million sequences, against the UniRef50 database, which contains approximately 40 million sequences.[15] The search took 5.42 hours and performed 23.0 billion alignments in the very sensitive mode and 17.77 hours while performing 23.1 billion alignments in the ultra-sensitive mode. The ExaBiome paper notes that this translates to 1.2 million sequences per second evaluated in a search space of 281 × 39 × 1012 sequences.
To demonstrate the value of their innovative (and transformative) approach for the Gordon Bell contest, the ExaBiome team performed an all-to-all comparison on the largest publicly available dataset containing 405 million proteins using 3364 nodes (or 73%) of the Summit supercomputer. The search completed in approximately 3.4 hours and achieved 690.6 million sequences per second in evaluating a search space of 405 × 405 × 1012 sequences.
When addressing this significantly larger problem, the ExaBiome team reported that they achieved a faster run time (PASTIS in 3.4 hours and DIAMOND in 5.42 hours), which is a remarkable achievement because PASTIS had to search a worst-case scenario O(N2) larger search space compared to the DIAMOND query. The numbers tell the story with theoretical pairwise comparisons by PASTIS at 405 × 405 × 1012 and by DIAMOND at 281 × 39 × 1012. This is where the GPU acceleration and intelligent use of sparsity for work savings really paid a run time dividend for the ExaBiome team—an investment that will deliver significant speedups for research scientists in the future.
Sensitivity and Search Space
Of course, the preferred method is to compare DIAMOND’s sensitivity and run time by using the exact same, larger dataset.
Because this was not possible, the team instead scaled the results reported by the DIAMOND team[16] to match the search space of the experiment performed on the Summit supercomputer.
Assuming DIAMOND exhibits perfect linear scaling to run on 2,025 Cora computational nodes (as opposed to the 520 nodes used in the DIAMOND paper), the ExaBiome team projected it would take DIAMOND 12.53 hours to evaluate the same search space that PASTIS examined in 3.44 hours. According to this analysis, the ExaBiome many-to-many search is 3.6× faster.
Based on the greater number of pairwise alignments performed, the ExaBiome team also claims a 24.8× increase in sensitivity compared to the projected equivalent DIAMOND run.Consult the paper for a more detailed discussion.
An End-To-End Scalable Protein Family Detection Pipeline
PASTIS and HipMCL form an end-to-end scalable protein family detection pipeline (Figure 2).
In this pipeline, PASTIS forms the protein similarity graph from raw sequences, and HipMCL clusters the graph into protein families by using the same HPC hardware resources. Protein families are sets of homologous proteins across all species that evolved from a common ancestor and likely perform similar functions. By efficiently discovering protein families from metagenomic datasets, the PASTIS + HipMCL pipeline empowers biologists to make rapid discoveries of new protein families that are related for some structural or functional reason and might have previously unknown yet potentially significant functionality.
The computationally intensive pairwise alignment operations form the main computational bottleneck. PASTIS is a unique solution in that it exploits the power of distributed GPU accelerators to intelligently address the runtime growth of the many-to-many pairwise comparisons. Thus, PASTIS also provides scientists with access to the latest generation of exascale supercomputers, all of which rely on GPUs to deliver extreme performance.
Furthermore, the pipeline takes advantage of the heterogeneous architecture of GPU-accelerated systems to hide the overhead of the distributed overlap detection component of the search by performing them on the CPUs simultaneously with the alignment operations on the accelerators. This also leverages the performance of both the CPU and GPU memory subsystems as overlap detection is a memory-bound operation.
To prevent I/O from becoming a bottleneck, Buluç notes, “We utilized a modified version of the distributed 2D Blocked Sparse Scalable Universal Matrix Multiplication Algorithm (SUMMA) algorithm that allows us to perform the search incrementally. This is important as the scale of the search increases because with it we can effectively control the maximum amount of memory required by the entire search. Further, our approach only performs I/O at the beginning and at the end of the search. Both are performed in parallel and constitute at most 3% of the entire search time.”
Buluç continues, “We also rely on custom load balancing techniques and distributed sparse matrices as the founding structures, the parallel performance of which is well-studied in numerical linear algebra. Overall, we obtain good scalability by attaining more than 75% strong-scaling and 80% weak-scaling parallel efficiency.” This scaling behavior is important to scientists because it means they can efficiently utilize more computational resources on larger machines to analyze larger datasets.
The type of load balancing employed depends on the form of the overlap matrix, which is a sparse matrix in which each nonzero entry represents a pairwise alignment that must be performed.
The overlap matrix is created by the 2D Sparse SUMMA algorithm. The matrix is symmetric because the similarity graph is undirected, which is a characteristic leveraged by the team in proposing two load balancing schemes designed to save on run time.
- Triangle-based load balancing: Only the blocks with a non-empty intersection with the strictly upper triangular portion of the overlap matrix are computed.
- Index-based load balancing: All the blocks are computed and pruned to preserve the original nonzero distribution of the overlap matrix, which is usually uniform.
The strong-scaling behavior on Summit with each of the load balancing schemes is shown in Figure 6.
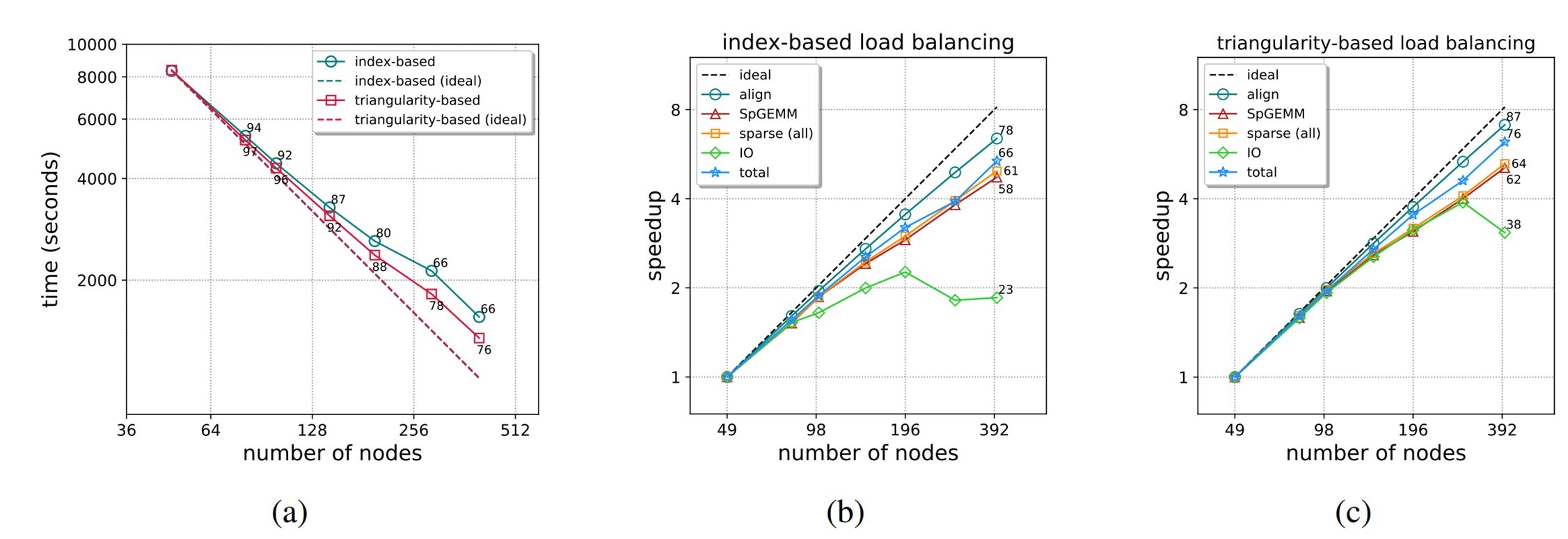
Figure 6. Strong-scaling performance. The annotated values in the plots indicate the attained parallel efficiency (%).
The weak-scaling parallel efficiency of the different components is reported in Figure 7. The green line highlights the impact of I/O on performance as the number of nodes increases. The blue line with a star shows the total efficiency, which remains high as the number of nodes increases.
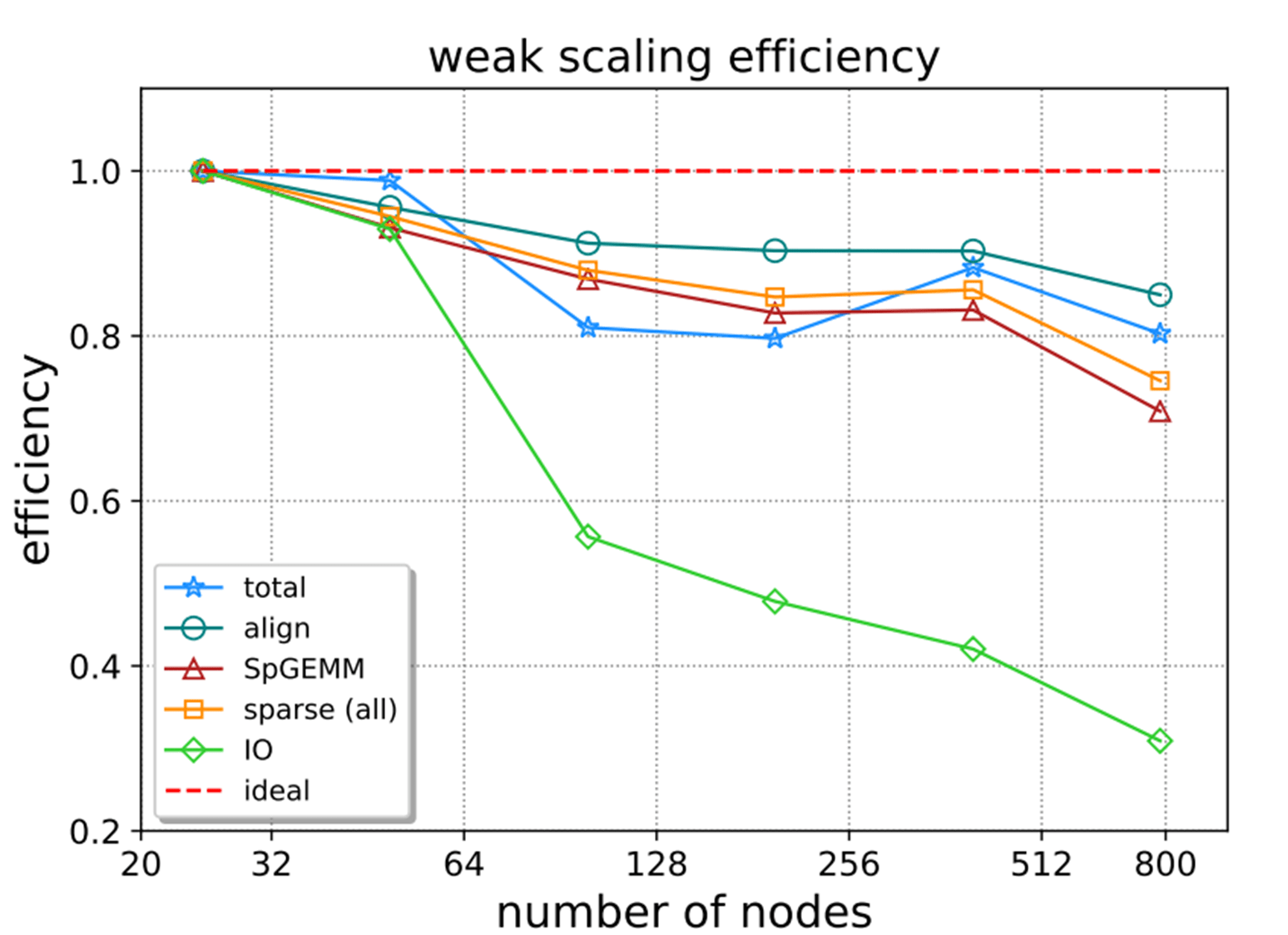
Figure 7. Weak-scaling of different components.
Harnessing HPC Resources for Biological Insight
The ExaBiome project harnesses HPC resources to go from raw metagenomic sequencing data to biological insight. Other software utilized by ExaBiome, such as MetaHipmer, can take raw metagenome data and assemble it to produce longer sequences on which protein coding regions can be discovered and translated. PASTIS + HipMCL takes those proteins to discover new protein families with distinct functionalities. Hence, ExaBiome provides an integrated pipeline for analyzing newly sequenced metagenomic datasets on systems—be they CPU only or GPU accelerated.
According to the paper, “The key to high performance, as we demonstrate in our work, is the good orchestration of on-node and node-level parallelism with techniques that can overcome the performance bottlenecks.”
This research was supported by the Exascale Computing Project (17-SC-20-SC), a joint project of the U.S. Department of Energy’s Office of Science and National Nuclear Security Administration, responsible for delivering a capable exascale ecosystem, including software, applications, and hardware technology, to support the nation’s exascale computing imperative.
Rob Farber is a global technology consultant and author with an extensive background in HPC and in developing machine learning technology that he applies at national laboratories and commercial organizations.
[1] https://sites.google.com/lbl.gov/exabiome/presentations
[2] https://www.nature.com/articles/s41467-018-04964-5
[3] Extreme-scale many-against-many protein similarity search Oğuz Selvitopi, Saliya Ekanayake , Giulia Guidi, Muaaz G. Awan , Georgios A. Pavlopoulos, Ariful Azad , Nikos Kyrpides, Leonid Oliker , Katherine Yelick, Aydın Buluç
[4] ibid
[5] ibid
[6] ibid
[7] https://pubmed.ncbi.nlm.nih.gov/11917018/
[8] https://academic.oup.com/nar/article/46/6/e33/4791133
[9] https://docs.olcf.ornl.gov/systems/crusher_quick_start_guide.html
[10] See note 3
[11] https://people.eecs.berkeley.edu/~aydin/GALLA-sparse.pdf
[12] https://www.exascaleproject.org/publication/adept-introduced-to-improve-large-scale-bioinformatics-data-analysis/
[13] https://www.unmc.edu/bsbc/education/courses/gcba815/Week3_Blast.pdf
[14] https://www.nature.com/articles/s41592-021-01101-x
[15] ibid
[16] ibid